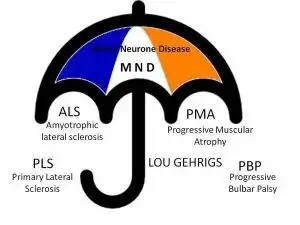
Motor Neurone Disease - MND
Amyotrophic Lateral Sclerosis (ALS) and other MND diseases
Motor Neurone Disease (MND) is a collective name for muscle diseases that affect the motor nerve cells. Motor nerve cells transmit information from the brain to the muscles.
MND includes:
- Amyotrophic Lateral Sclerosis (ALS) or (Lou Gehrig's disease)
- Progressive Spinal Muscular Atrophy (PSMA)
- Primary Lateral Sclerosis (PLS)
- Progressive Bulbar Palsy (PBP)
Source:
ALS
ALS is a serious disease of motor nerve cells in the central nervous system (brain and spinal cord) and the peripheral nervous system. The peripheral nervous system provides a connection between the brain and spinal cord (the central nervous system) and the muscles and organs. The nerve cells are needed to make movements. Because the nerve cells are affected, the brain cannot properly transmit signals to the muscles. As a result, the brain can no longer control the muscles to make a movement.
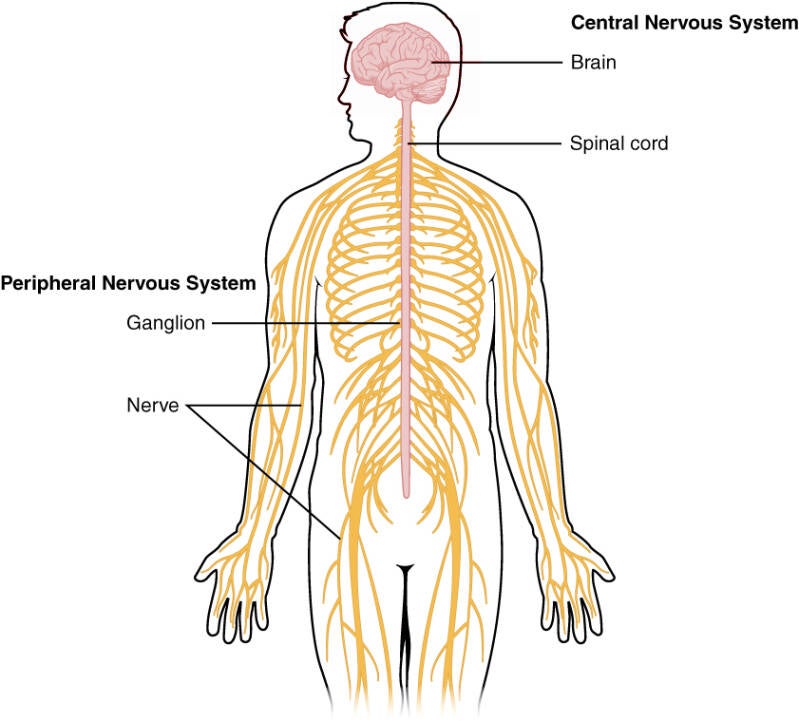
Image from: https://commons.wikimedia.org/w/index.php?curid=30147907
The pink color is the central nervous system and the yellow color is the peripheral nervous system.
ALS is a progressive disease. This means that it is a chronic disease that becomes increasingly serious. Because more and more nerve cells either do not function fully or do not work at all, muscle atrophy begins to occur. This means that the muscles become smaller and thinner. Because the disease is progressive, the nerve cells are increasingly affected and this leads to increasing muscle weakness. Once the nerve cells are severely or even completely affected, the patient can become paralyzed. This entire process causes various limitations in functioning.
Little is known about the cause. It has been found that in patients with ALS, just like in Parkinson's patients, there is too much glutamate in the nervous system. Glutamate is a neurotransmitter (a type of protein) that is important for signal transmission from one nerve cell to another. Once the signal has been transmitted, the glutamate is normally broken down. In ALS, glutamate is not broken down properly or too much is released. As a result, glutamate accumulates in the nerve endings. This causes damage to the nerve cells.
In about 10% of cases, ALS is hereditary (familial variant), but in 90% of cases ALS is not hereditary (sporadic form).
Approximately 400 to 500 people in the Netherlands are diagnosed with ALS each year. On average, ALS starts between the ages of forty and sixty. The average life expectancy after diagnosis is about three years. This means that there are also exceptions where people live longer than three years after diagnosis.
Symptoms
- Movement/muscle relaxation
The first symptoms are often reduced strength in the arms or legs, difficulty speaking (dysarthria), swallowing and breathing. The increasing muscle weakness also causes spasticity and joint pain. As the patient becomes increasingly paralyzed, it becomes more difficult to change body position. This, and increased muscle tension (due to increasing spasticity) can cause a lot of pain.
A slight decline in thinking and behavior develops in approximately thirty percent to half of patients. This includes a reduction in empathy, reduced inhibition, memory loss and reduced speed of speech.
About 5 to 10% of patients have frontotemporal dementia.
This is a serious form of dementia in which, among other things, major behavioral changes occur. Loss of initiative, lack of insight, disinhibition and memory problems can also occur.
- Speaking and swallowing
Due to increasing deterioration of the tongue and jaw muscles, chewing and swallowing becomes more difficult.
Patients increasingly suffer from a swallowing disorder. This can lead to dehydration, malnutrition and a dry mouth, but also to too much saliva in the mouth.
In addition, food can more easily end up in the lungs, which can cause pneumonia.
- Fatigue/sleep problems
Due to anxiety and being constantly confronted with the disease, falling asleep can occur. Deteriorating respiratory muscles can also cause regular sleep interruptions.
Diagnosis
- Consultation with the GP or anotther doctor
Initially, a conversation will take place with the doctor. She / he will look at the complaints that are present and the medical history. A family tree study can also take place to see whether ALS or Frontotemporal Dementia (FTD) also occurs elsewhere in the family.
- Neurological examination
With a neurological examination, the neurologist looks at various functions. The patient's cranial nerves, strength, coordination, reflexes, senses and sensation are tested. Mental function is also tested thoroughly.
Muscle weakness, tremors under the skin, thinning of the muscles and increased reflexes are the most important symptoms of ALS.
- Blood test
The blood is tested to rule out certain diseases. The chemical and metabolic substances in the blood are used to estimate what kind of condition it may be.
- EMG test
Electromyography (EMG) measures electrical activity in the muscles and the activity of the nerve cells. The EMG can be used to determine whether enough signals are being sent and whether the muscles are weaker.
- Lung function test
This test is performed to test the respiratory muscles to see if they have enough strength to maintain breathing properly.
- MRI scan and/or muscle biopsy
In some cases, an MRI scan or muscle biopsy is performed to gain clarity about any abnormalities.
Treatment
ALS unfortunately cannot yet be cured.
However, a drug is used to slow down the disease process.
Riluzole is currently the only drug that is used. Riluzole slows down the disease process, but cannot cure the disease. It is still unknown how the drug Riluzole works exactly for ALS. It is known that Riluzole blocks glutamate and thus possibly slows down the damage to the nerve cells.
Research
ALS cannot yet be cured and the disease and disease process are rapid. More research is still needed to unravel the cause and the functioning of ALS. Research into possible medicines to treat and/or cure ALS is also urgently needed. Several studies are already underway and experimental trials are being conducted.
For ongoing research, see:
The Netherlands: www.als.nl/onderzoek/onderzoeksprojecten/
USA: https://www.als.org/research
Other MND diseases
As mentioned earlier on this page there are, besides ALS, several forms of Motor Neurone Disease (MND).
PSMA (Progressive spinal muscular atrophy)
PSMA involves problems with the peripheral motor nerve cells, not with the brain or spinal cord. PSMA manifests itself in the muscles becoming weaker and thinner. With the exception of spasticity, all symptoms that also occur with ALS can occur. PSMA has two types, a rapidly progressive and a slowly progressive variant. In some patients, PSMA can progress to ALS after months or years.
PLS (Primary Lateral Sclerosis)
PLS is a disease of the central motor nerve cells, specifically in the brain and spinal cord. PLS is characterized by stiffness, increased muscle tension, increased reflexes and muscle weakness.
Usually this disease starts in the legs, later the arms/hands and this will worsen with age. Symptoms are very similar to those of ALS, such as chewing and swallowing problems and difficulty speaking.
PLS has a slower course than ALS. People can live with PLS for decades. In some patients, PLS can turn into ALS after years. If the disease lasts longer than four years, the chance of this is smaller.
PBP (Progressive bulbar palsy)
PBP is a disease of nerve cells in the brain stem (the medulla oblongata, also known as the bulbus cerebri).
Because the disease is progressive, nerve cells function less and less well, which leads to increasing paralysis (paresis) of the muscles of the mouth, tongue and throat (bulbar).
The cranial nerves that supply the bulbar muscles are the glossopharyngeal nerve (IX), vagus nerve (X) and hypoglossal nerve (XII). See here for information about the cranial nerves.
Characteristic are complaints that manifest themselves in difficulty chewing, swallowing and speaking. Involuntary laughing, crying or yawning can also occur, as well as weakness of the facial muscles.
The chance of pneumonia due to choking (aspiration pneumonia) is high because The patient can choke or accidentally get food or drink in the airway and lungs when inhaling.
PBP can progress to ALS. The muscle weakness then also manifests itself in the arms and/or legs.
Resources
Team hersenletsel-uitleg
www.als.be/nl/ALS-onderzoek/
www.als-centrum.nl/kennisplatform/riluzole
www.apotheek.nl/medicijnen/riluzol
www.erfelijkheid.nl/ziektes/als-amyotrofische-lateraal-sclerose
Foran, Emily, and Davide Trotti."Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis." Antioxidants & Redox Signaling, vol. 11, no. 7, 2009.
www.gezondheidenwetenschap.be/richtlijnen/amyotrofe-laterale-sclerose-als
www.hersenletsel-uitleg.nl/soorten-hersenletsel-hersenaandoeningen/degeneratieve-aandoening/ftd
www.spierziekten.nl/overzicht/amyotrofische-laterale-sclerose/diagnose-en-verschijnselen-als/
www.umcutrecht.nl/nl/ziekenhuis/ziekte/als
Hughes TAT and Wiles CM. Neurogenic dysphagia: the role of the neurologist. J Neurol Neurosurg Psychiatry. 64:569-572. (1998